
Om een geneesmiddel voor mensen op de markt te kunnen brengen, heeft een farmaceutisch bedrijf een handelsvergunning nodig. Er zijn twee soorten handelsvergunningen die kunnen worden aangevraagd voor een geneesmiddel: een nationale handelsvergunning en een Europese handelsvergunning.
Er zijn drie verschillende procedures via welke een handelsvergunning kan worden verkregen:
- via een nationale procedure
- via een decentrale procedure of wederzijdse erkenningsprocedure
- via een centrale procedure
Een farmaceutisch bedrijf mag zelf kiezen welke procedure het wil volgen, tenzij het om een geneesmiddel gaat dat op grond van Europese wetgeving verplicht via de centrale procedure geregistreerd moet worden, zoals medicijnen om kanker te behandelen.
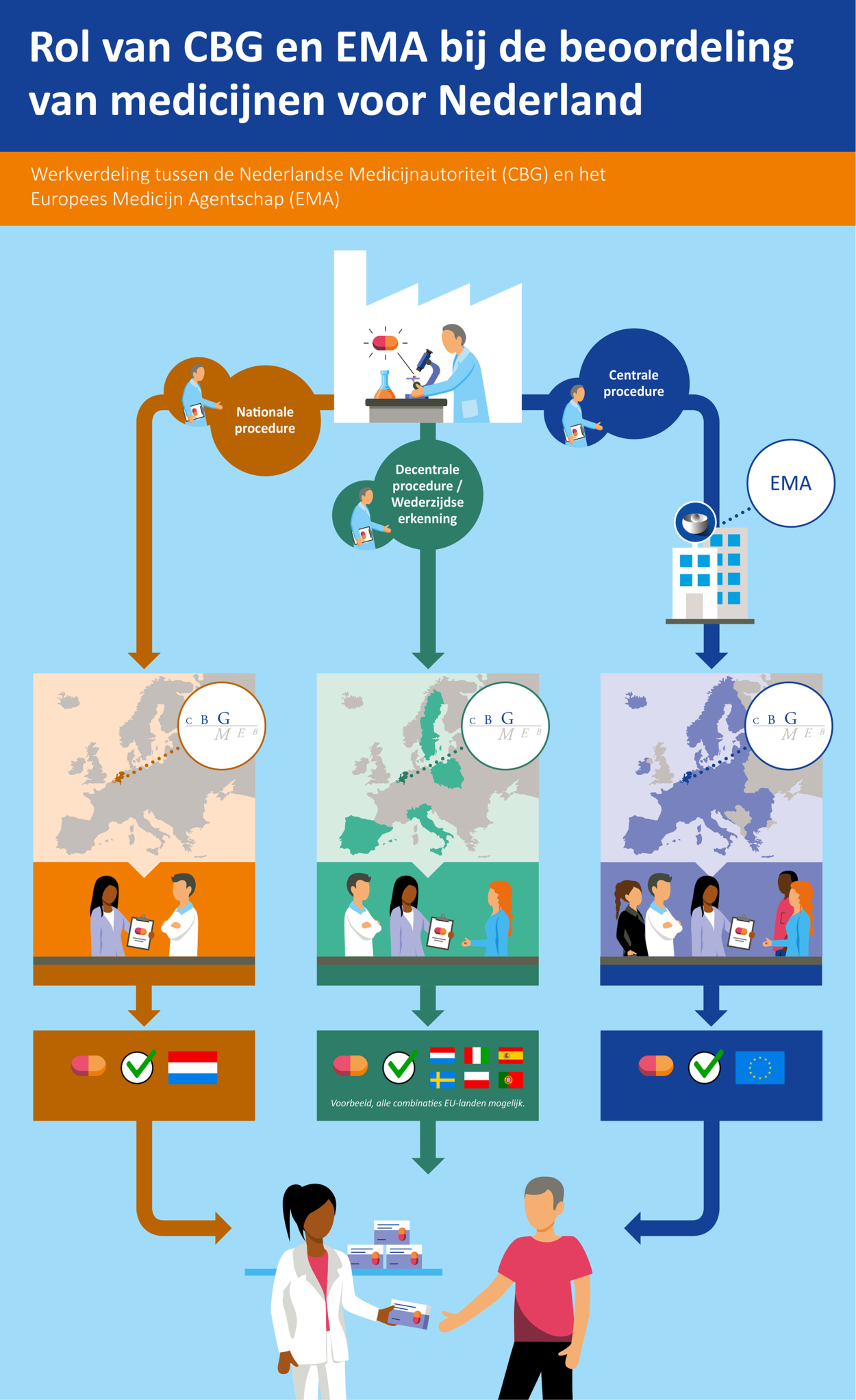
De nationale procedure
Bij de nationale procedure dient een firma een registratiedossier in bij de geneesmiddelenautoriteit van één lidstaat. In Nederland is dat het College ter Beoordeling van Geneesmiddelen (CBG). Het CBG beoordeelt het geneesmiddel op kwaliteit, werkzaamheid en veiligheid. Bij een positief oordeel verstrekt het CBG een handelsvergunning die alleen geldig is voor Nederland. Het geneesmiddel mag dan alleen in Nederland op de markt komen.
De decentrale procedure
Bij de decentrale procedure vraagt een firma de registratie van een geneesmiddel in meerdere lidstaten binnen de Europese Unie (EU)* tegelijkertijd aan. Deze procedure is alleen mogelijk als het geneesmiddel in nog geen enkele Europese lidstaat een handelsvergunning heeft gekregen. De firma vraagt één land om referentieland in de procedure te worden, bijvoorbeeld Nederland. Dit land is dan de Reference Member State (RMS). Deze referentielidstaat leidt de beoordeling van het registratiedossier. De andere betrokken lidstaten kunnen hun inbreng leveren in de beoordelingsprocedure.
De wederzijdse erkenningsprocedure
Wanneer een middel al een handelsvergunning heeft verkregen via een nationale of decentrale procedure, kan een bedrijf de registratie ook uitbreiden naar andere EU-landen. Dit gaat via de wederzijdse erkenningsprocedure. De geneesmiddelenautoriteit die de oorspronkelijke beoordeling gedaan heeft (de RMS), houdt de leiding bij deze procedure.
Bij de decentrale en wederzijdse erkenningsprocedure volgt na de goedkeuring van een geneesmiddel een aparte nationale fase. In die fase worden door de firma de Nederlandse vertalingen gemaakt van onder andere de bijsluiter en geeft het CBG vervolgens, na beoordeling van de vertalingen, op nationaal niveau een vergunning af.
De centrale procedure
Bij de centrale procedure is de toelating van het geneesmiddel in één keer geldig voor de gehele Europese Unie (EU). Bij deze procedure wordt het registratiedossier door een farmaceutisch bedrijf ingediend bij het Europees Medicijnagentschap (EMA). Het dossier wordt beoordeeld door twee lidstaten: de lidstaat die de rapporteur levert en de lidstaat die de co-rapporteur levert. Deze (co‑)rapporteurs hebben namens hun land zitting in het Committee for Medicinal Products for Human Use (CHMP), waarin alle lidstaten zijn vertegenwoordigd. Na bespreking van de rapporten die door de (co-)rapporteurs zijn opgesteld, stuurt de CHMP zijn eindoordeel naar de Europese Commissie voor definitieve besluitvorming.
Het volgen van de centrale procedure is verplicht voor geneesmiddelen die biotechnologisch bereid worden en voor nieuwe geneesmiddelen die bedoeld zijn voor de behandeling van onder andere kanker, aids, neurodegeneratieve ziekten, diabetes en virale ziektes (zoals COVID-19). Ook geneesmiddelen in het kader van gen- en celtherapie vallen onder deze verplichting. De reden van de verplichting is om patiënten in heel Europa toegang te geven tot deze geneesmiddelen.
Rolling review
In 2020 is er voor de beoordeling van verschillende coronavaccins en geneesmiddelen voor de behandeling van COVID-19 een versnelde procedure toegepast. Bij een zogenoemde ‘rolling review’ deelt een bedrijf al informatie met de medicijnautoriteiten zoals het CBG en het EMA, terwijl het klinische onderzoek nog in volle gang is. Zo kan de beoordeling eerder starten. Beoordelingen via rolling review leveren aanzienlijke tijdwinst op, zonder dat er concessies worden gedaan op de beoordeling van de kwaliteit, veiligheid en effectiviteit van het vaccin of geneesmiddel.

Vaccin Comirnaty van BioNTech/Pfizer in EU goedgekeurd tegen COVID-19
- Een mRNA-vaccin dat bestaat uit 2 prikken werkt bij gemiddeld 95 van de 100 mensen.
- Het vaccin zorg ervoor dat het lichaam antistoffen maakt tegen het coronavirus.
- Mogelijke bijwerkingen: roodheid, pijn of zwelling bij de prikplek, hoofdpijn, vermoeidhoeid en koorts.
- Voor jongeren vanaf 16 jaar, volwassenen en ouderen (65+). Zwangere vrouwen: raadpleeg uw arts of u dit vaccin mag krijgen.
Parallelimport
Een andere route om een geneesmiddel op de markt te brengen is via parallelimport. Hierbij wordt een geneesmiddel geïmporteerd uit een andere Europese lidstaat door een importeur. Het parallelproduct moet gelijk of nagenoeg gelijk zijn aan het Nederlandse referentiegeneesmiddel. Er mag geen verschil zijn in werkzaamheid en veiligheid. Het CBG beoordeelt of het parallelproduct uitwisselbaar is met een al in Nederland geregistreerd geneesmiddel.
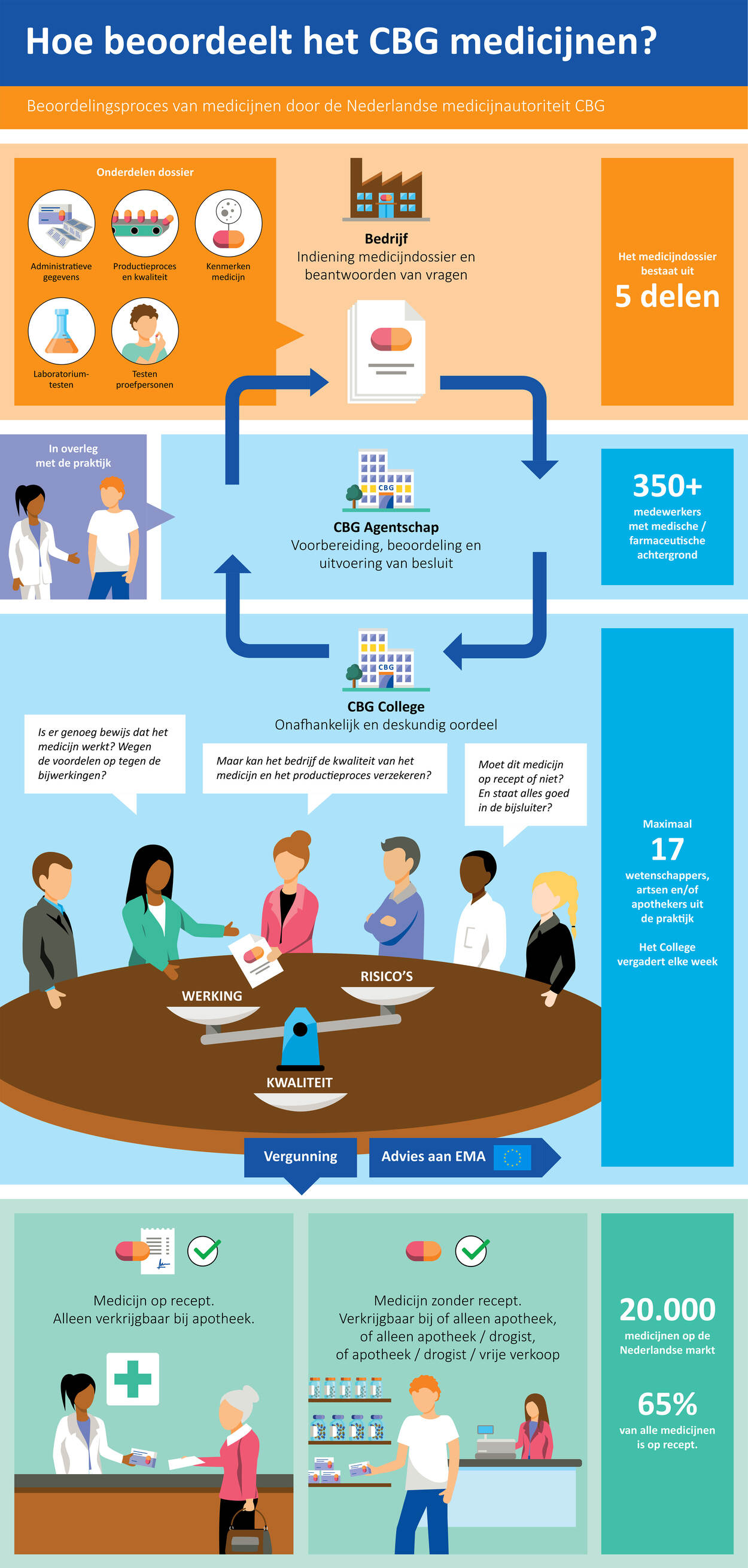
Hoe het CBG geneesmiddelen beoordeelt
Een farmaceutisch bedrijf mag zijn geneesmiddel voor mensen pas op de markt brengen na goedkeuring door een geneesmiddelenautoriteit. In Nederland is dat dus het CBG. Dat beoordeelt het geneesmiddel op werkzaamheid, veiligheid en kwaliteit. Zo’n beoordeling verloopt via een vaste procedure.
*Het Europese samenwerkingsverband bestaat uit de landen uit de Europese Unie aangevuld met IJsland, Noorwegen en Liechtenstein, de zogenaamde Europese Economische Ruimte (EEA). In het jaarverslag wordt hiernaar verder verwezen als ‘Europese Unie’, ‘EU’ of ‘lidstaten’.







